
Apart from the interactive execution mode, Jupyter Notebooks support a bulk execution mode, in which all the code cells are executed sequentially from the first to the last. Users can initiate a bulk execution through the Run All button in the web interface.
Jupyter Workflow enriches Notebooks’ bulk execution with a DAG-based execution mode, which aims to execute concurrently all those cells that do not depend on each other through true data dependencies. When dealing with complex workflows, the amount of concurrency allowed by the DAG-based execution mode becomes significant.
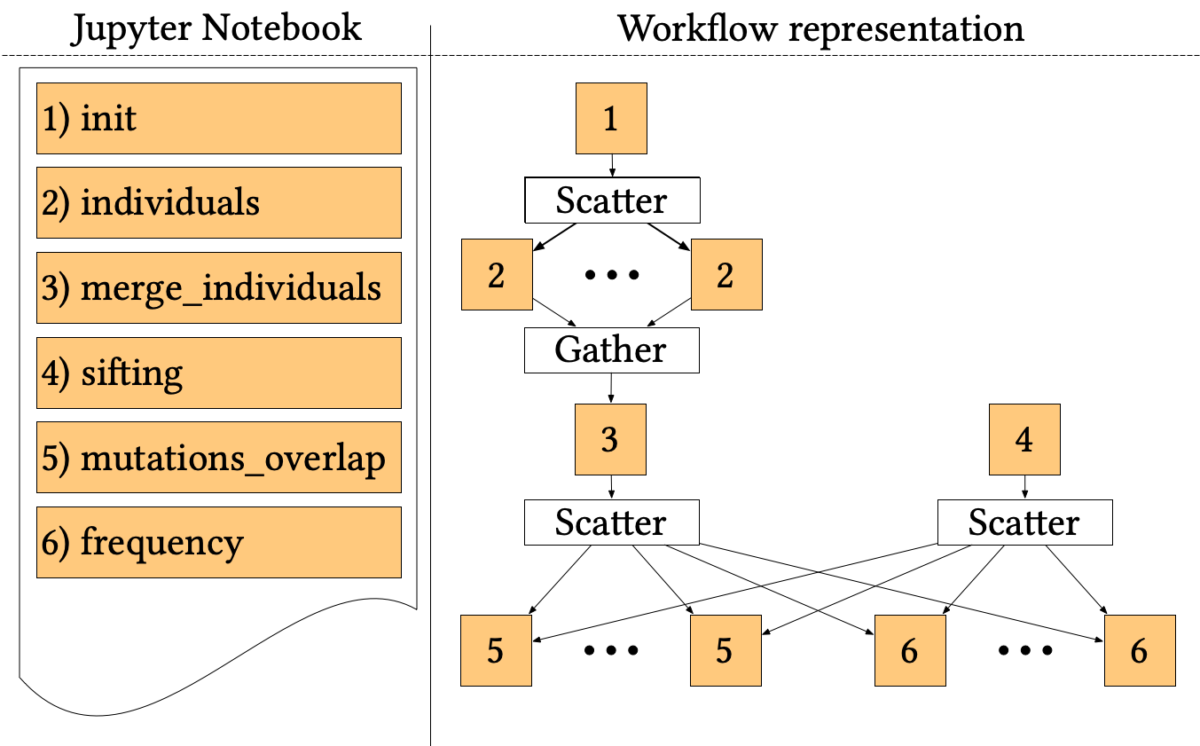
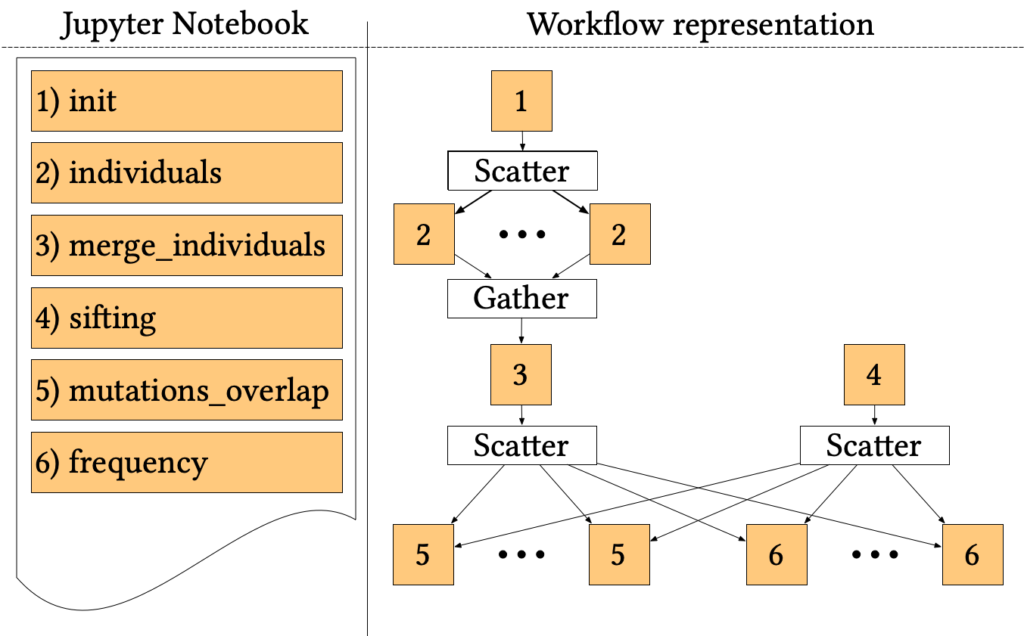
The 1000-genome workflow
The 1000-genome workflow is a Bioinformatics pipeline aiming at fetching, parsing and analysing data from the 1000 Genomes Project to identify mutational overlaps and, in particular, disease-related mutations. Initially implemented with the Pegasus Workflow Management System, it consists of five steps.
The first two steps fetch and parse the Phase3 data by chromosome. These files list all Single Nucleotide Polymorphisms (SNPs) variants in that chromosome and which individuals have each one. An individual step creates output files for each individual of rs numbers 3, where individuals have mutations on both alleles.
Since individuals’ files are huge, each file is divided into chunks, which are processed in parallel by the first step using a Scatter/Gather pattern. Then, the second step merges the processed fragments in a single mutations file.
A sifting task (step 3) computes the SIFT scores of all the SNPs variants, as calculated by the Variant Effect Predictor (VEP). The sifting task processes the corresponding VEP for each chromosome, selecting only the SNPs variants that have a SIFT score.
The last two steps analyse the mutations to extract valuable measures. In particular, step 5 measures the overlap in mutations among pairs of individuals by population and by chromosome. Step 6 calculates the frequency of overlapping in mutations by selecting several random individuals and all SNPs variants without considering the SIFT scores.
The entire pipeline, written in a mixture of Bash and Python, has been trivially ported to Jupyter Workflow, using a cell for each step and proper metadata to encode dependencies and parallel patterns. The resulting Notebook is available online at the official Jupyter Workflow GitHub repository.
Jupyter Workflow execution
The critical portion of the workflow is the first step, composed of 2000 independent short tasks (~120s each). These steps are critical for batch workload managers because the waiting time in the queue is comparable with the step execution time. Nevertheless, they can still be executed at scale on on-demand Cloud resources (e.g. Kubernetes).
We measured the strong scaling of an 8-chromosomes instance of the workflow for performance evaluation on 500 concurrent Kubernetes Pods. In particular, we focused on step 1, which is the bottleneck of the entire pipeline.
We deployed a Kubernetes cluster with 3 control plane VMs (4 cores, 8 GB RAM each) and 16 large worker VMs (40 cores, 120 GB RAM each) interconnected with a 10 Gbps Ethernet technology. Each Pod requests 1 core and 2 GB of RAM, and mounts a 1 GB tmpfs.
We also developed a DryRun version of the code, serving as a baseline. The DryRun simulates the workflow behaviour without using CPU cores and network bandwidth: the business code is replaced with sleep instructions of the expected average timespan of the task, and communications are replaced with small messages.
The actual workflow scaled reasonably well up to 250 containers. Then, it started suffering from the Kubernetes master bottleneck for data distribution. In contrast, the DryRun implementation shows that the intrinsic overhead introduced by Jupyter Workflow runtime synchronisations keeps a reasonably linear gap against ideal speedup, at least up to 500 Pods.
I. Colonnelli, M. Aldinucci, B. Cantalupo, L. Padovani, S. Rabellino, C. Spampinato, R. Morelli, R. Di Carlo, N. Magini and C. Cavazzoni, “Distributed workflows with Jupyter”, Future Generation Computer Systems, vol. 128, pp. 282-298, 2022. doi: 10.1016/j.future.2021.10.007